2026 Mentors: Ratul Chowdhury
The input to most computational chemistry packages is Cartesian coordinates of a molecule (i.e., the location of each atom in three-dimensional space). For example the Cartesian coordinates of a water dimer are (units are Angstroms, i.e., one tenth of a nanometer):
H -1.958940 -0.032063 0.725554
H -0.607485 0.010955 0.056172
O -1.538963 0.004548 -0.117331
H 1.727607 0.762122 -0.351887
H 1.704312 -0.747744 -0.399151
O 1.430776 -0.003706 0.113495
Interestingly, using the laws of physics, this is almost all of the information that we need in order to compute the interactions in this system. However, as chemists, we have developed our intuition not based on where atoms in space are, but on how atoms are connected, i.e., which atoms are bonded.
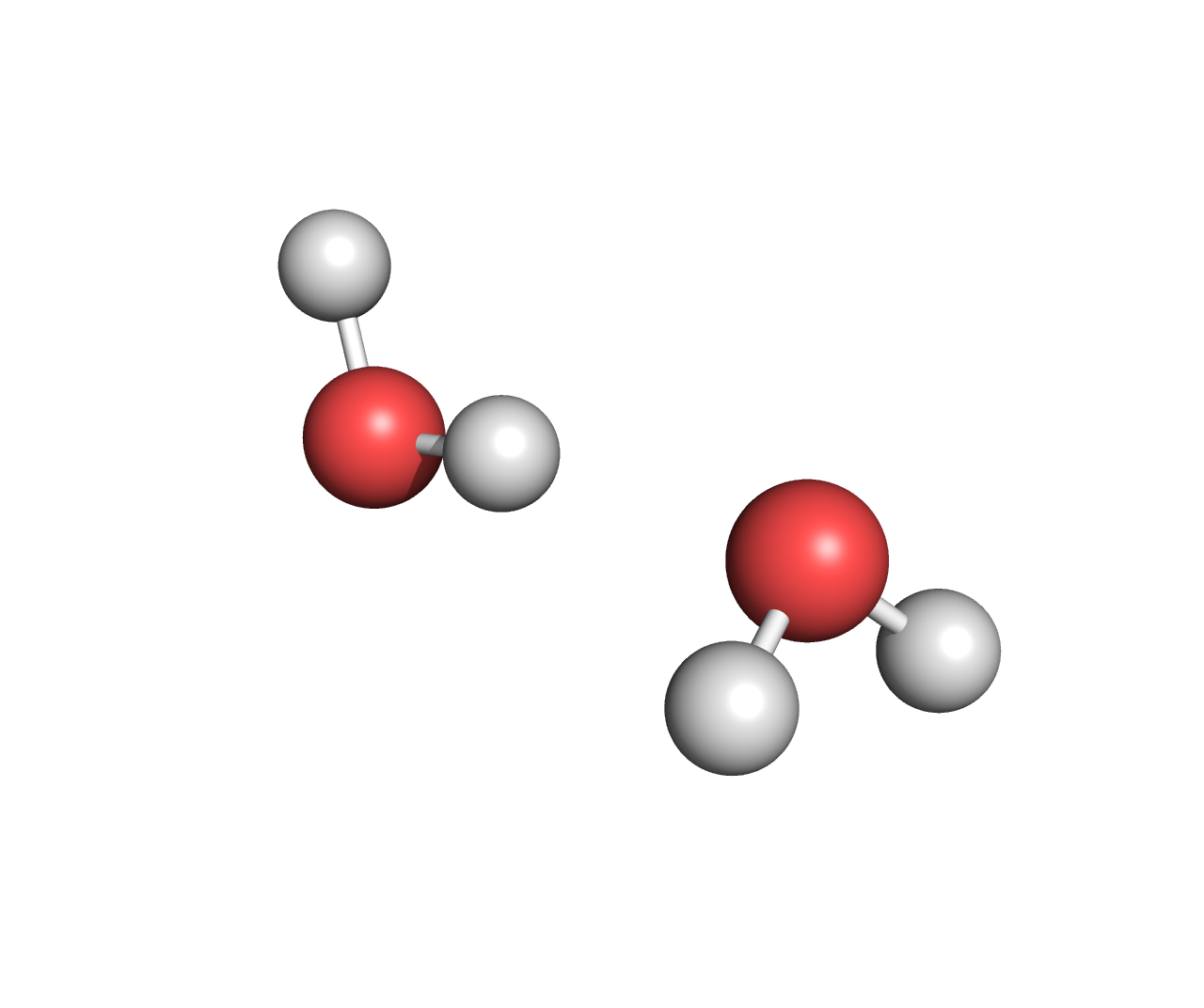
As chemists, we can plot the water geometry above and quickly assign bonds by visual inspection. Our intuition immediately tells us the first two hydrogens are covalently bonded to the first oxygen, the second two hydrogens are covalently bonded to the second oxygen, and the second hydrogen is hydrogen bonded to the second oxygen. Unfortunately, computers lack this intuition.
If we want a computer to make decisions based on the bonding of a chemical system, we must tell the computer where the bonds are. While we can easily specify the bonds in the water dimer, manually specifying where all the bonds in a protein are is extremely tedious and error-prone. We thus need to develop and rely on algorithms which can automatically assign the bonds.
Naively, one would assume this can be done simply by computing the distance between atoms, but what you’ll find is that bonds actually span a range of distances and there is not clear demarcations for when a distance falls within the bonding range, or for when it falls just outside that range.
This project seeks to apply machine learning to the problem.